
Reconciling Membrane Protein Simulations with Experimental DEER Spectroscopy Data
BibTeX
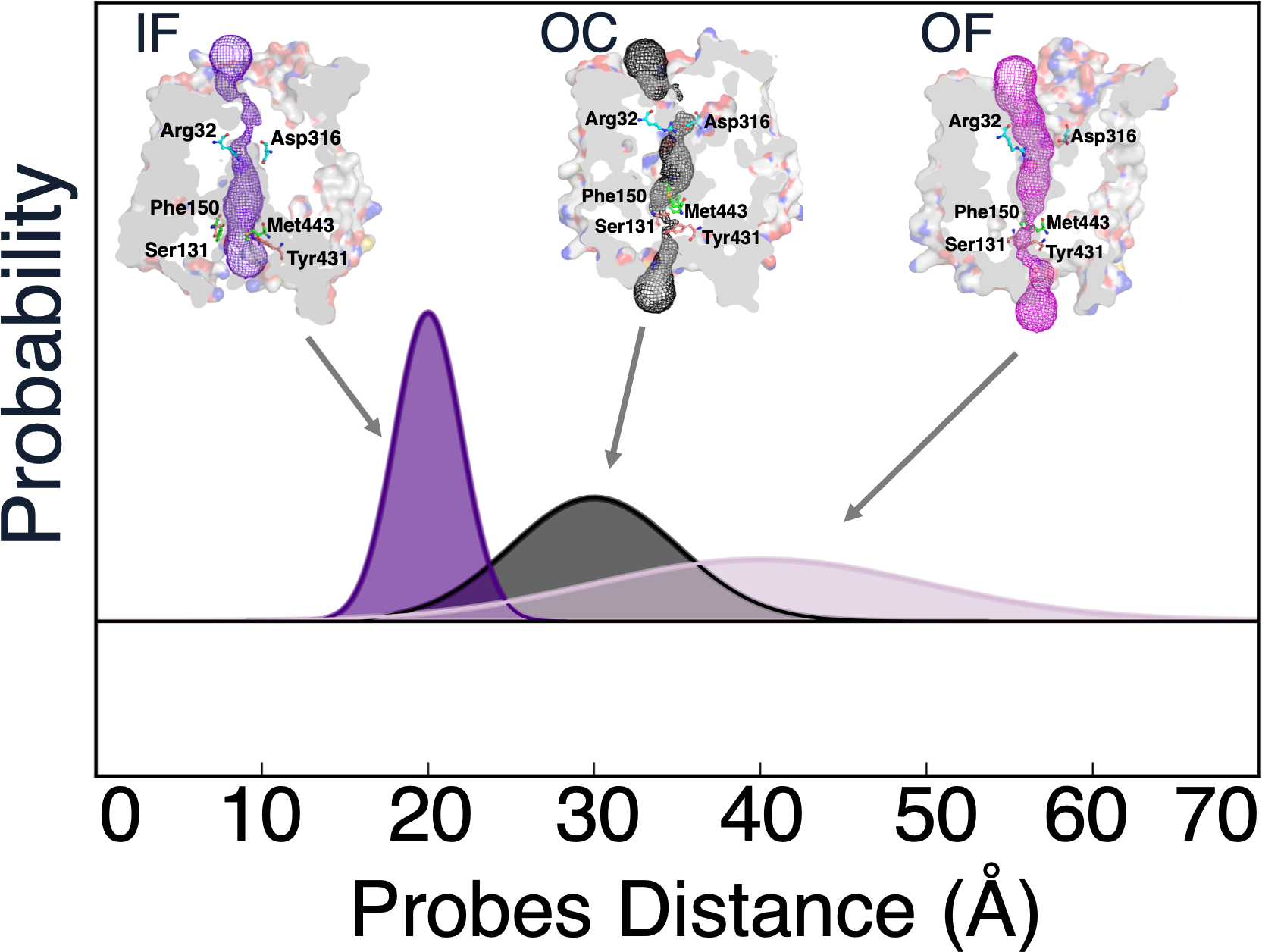
Spectroscopy experiments are crucial to study membrane proteins for which traditional structure determination methods still prove challenging. Double electron-electron resonance (DEER) spectroscopy experiments provide protein residue-pair distance distributions that are indicative of their conformational heterogeneity. Atomistic molecular dynamics (MD) simulations are another tool that have been proven to be vital to study the structural dynamics of membrane proteins such as to identify inward-open, occluded, and outward-open conformations of transporter membrane proteins, among other partially open or closed states of the protein. Yet, studies have reported that there is no direct consensus between the distributional data from DEER experiments and MD simulations, which has challenged validation of structures obtained from long-timescale simulations and using simulations to design experiments. Current coping strategies for comparisons rely on heuristics, such as mapping the nearest matching peaks between two ensembles or biased simulations. Here we examine the differences in residue-pair distance distributions arising due to the choice of membranes around the protein and covalent modification of a pair of residues to nitroxide spin labels in DEER experiments. Through comparing MD simulations of two proteins, PepTSo and LeuT–both of which have been characterized using DEER experiments previously–we show that the proteins’ dynamics are similar despite the choice of the detergent micelle as a membrane mimetic in DEER experiments. On the other hand, covalently modified residues show slight local differences in their dynamics and a huge divergence when the oxygen atom pair distances between spin labeled residues are measured rather than protein backbone distances. Given the computational expense associated with pairwise MTSSL labeled MD simulations, we examine the use of biased simulations to explore the conformational dynamics of the spin labels only to reveal that such simulations alter the underlying protein dynamics. Our study identifies the main cause for the mismatch between DEER experiments and MD simulations and will accelerate the development of potential mitigation strategies to improve the match.